Mass Fingerprinting using Tryptic Peptides.
When applied to a mixture of polypeptides derived from a protein spot, a mass spectrometry analysis yields a complex series of molecular masses, each of which corresponds to that of a peptide obtained from the trypsin digestion. Since it is assumed that the protein spot in the 2D gel was well isolated, it follows that all the measured peptides are derived from a single protein. Therefore, since trypsin specifically cleaves peptide bonds at lysine and argenine residues, the resulting peptides will have masses that are specifically related to the full protein sequence from which they were derived. The mass spectrum will therefore give a characteristic profile of peptide masses for a determined protein, and is commonly referred to as the "mass fingerprint" of a protein. Although at this stage there is no way of knowing which polypeptide gives rise to which peak in the mass spectrometry data, this data set is very useful when combined with genomic DNA sequence information.
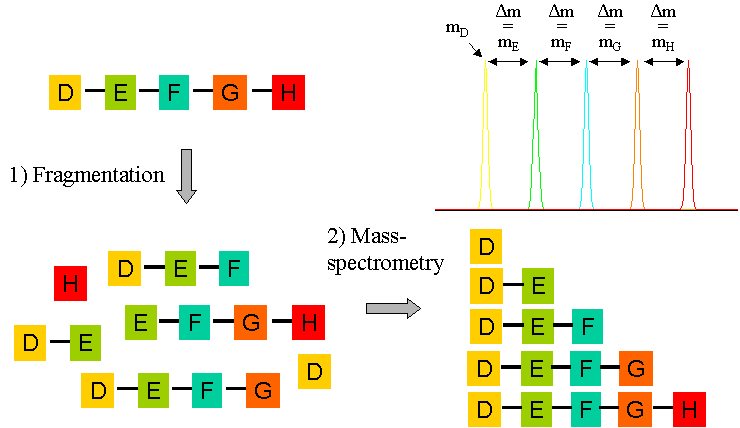
Modern mass spectrometers can go one step further in peptide analysis. The instrument can be tuned to analyse one particular mass peak (ie one chosen peptide), which is allowed to collide with an inert gas (usually argon). This results in the fragmentation of the peptide into the charged polypetide derivatives, and the differences in the masses of these derived "daughter ions" (mD, mE etc in the figure below) can be analysed to derive the amino acid sequence of the chosen peptide. In this way, not only can the mass of the peptide be found, but also its amino acid sequence. This peptide mass and sequence information is all that is needed for a comparison with the protein databases to identify the protein. This comparison works best when the protein database is derived from a genomic DNA sequencing project of the same organism.
One of the benefits of the rapid expansion in the area of genomics is the avalability of hundreds of whole genome sequences. Although the ability to analyse and interpret this data is barely keeping pace with the DNA sequencing technology, the number of reliably annotated genomes increases daily. An important part of genome annotation involves the identification of the open reading frames of all encoded proteins, and relies heavily on sophisticated (although not infallible) DNA sequence analysis algorithms. Since the predicted protein sequnces will be compared with mass spectrometry data, reliable and complete genome annotation is as important to the success of a proteomic project as the quality of the experimentally derived peptide mass data.
The positions of lysine and argenine residues in the amino acid sequence of a predicted protein may be located, and so the theoretical masses of the peptides resulting from and in silico tryptic digestion of this protein can be calculated. This analysis can rapidly be performed for all the predicted proteins in a genome, and the results compared to the peptide mass and peptide sequence data obtained by mass spectrometry using a sample derived from a single spot in the 2D gel. In this way, the measured and predicted mass fingerprints can be matched, and the protein in a given spot in the 2D gel can be identified.